CONTROL
EBGLYSS®, powerful skin improvement in monotherapy
ADvocate 1 (N=424) EBGLYSS® Monotherapy at Week 16
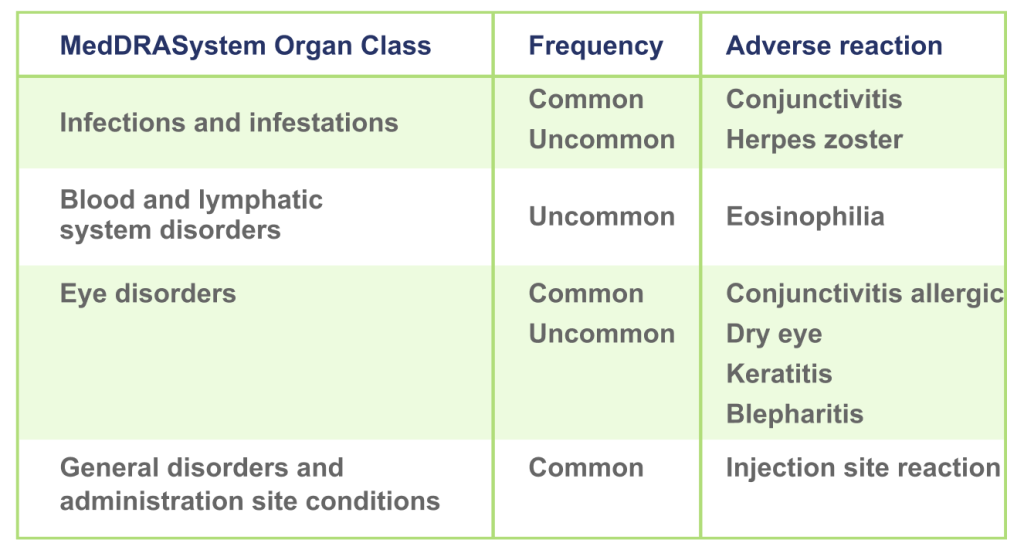
58.8% of adult and adolescent patients achieved EASI-75 at Week 161-2
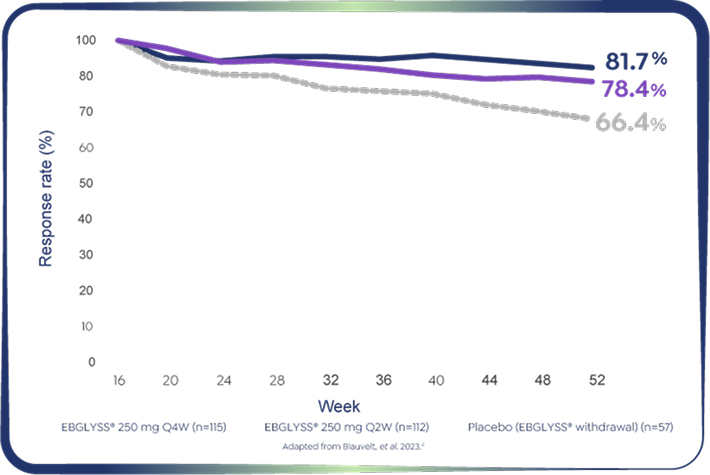
Pooled ADvocate 1&2: EBGLYSS® Monotherapy at Week 52
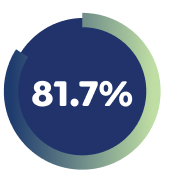
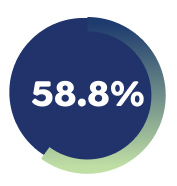
81.7% of Week 16 responders maintained EASI-75 with EBGLYSS® monotherapy at week 52 with Q4W.4
For the 36-week maintenance period, patients who received systemic rescue medication, discontinued treatment due to lack of efficacy or transferred to the escape arm had values set to their baseline value subsequent to this time through to Week 52; patients who received topical rescue medication or discontinued treatment for any other reasons had values set to missing. MCMC-MI was used to handle missing data.
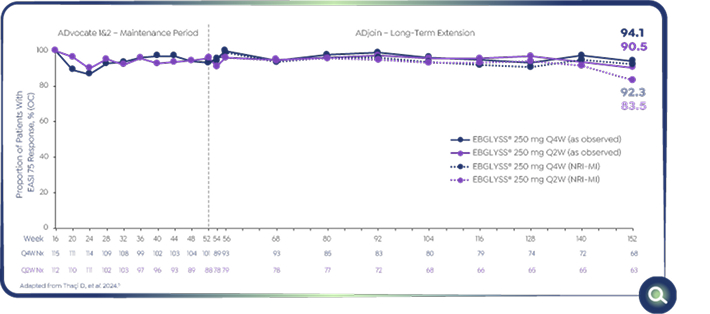
Pooled ADvocate 1&2 and ADjoin study: EBGLYSS® monotherapy at Week 152
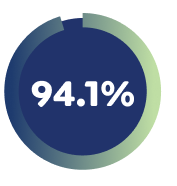
94.1% of Week 16 responders maintained EASI-75 with EBGLYSS® Q4W monotherapy at Week 152 (as observed)5
Efficacy analyses:
-As -observed (OC) analyses used all collected data regardless of rescue medication use.
-In addition to as observed analyses, the non-responder imputation-multiple imputation (NRI-MIc) method was implemented to handle missing data. For each imputation process, 25 datasets with imputations were calculated using SAS® software version 9.4.5
EBGLYSS®, meaningful relief from intolerable itch
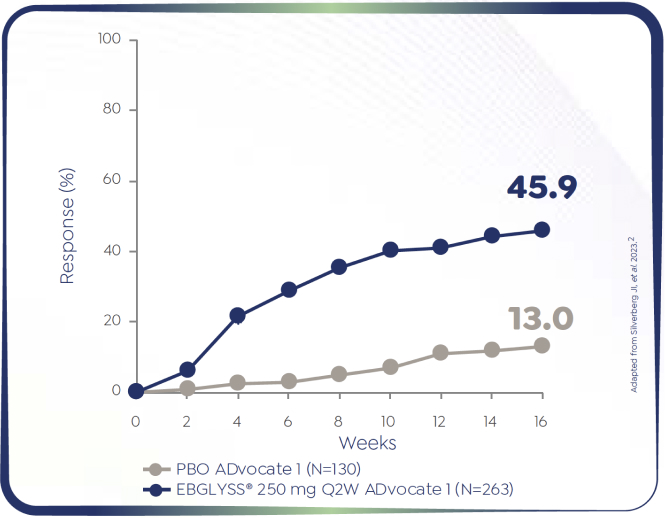
ADvocate 1 (N=424) EBGLYSS® monotherapy at Week 16
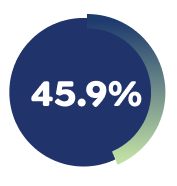
84.7% of Week 16 responders had achieved a Pruritus NRS ≥4-point improvement with EBGLYSS® monotherapy at Week 52 with Q4W.4
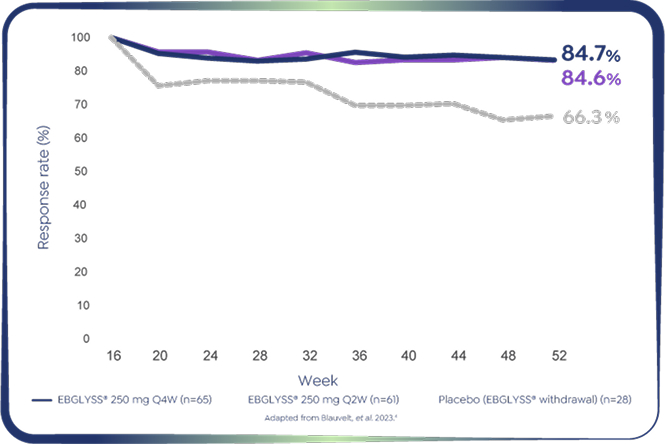
Pooled ADvocate 1&2: EBGLYSS® Monotherapy at Week 52
Percentage of patients who were EBGLYSS® resonders at Week 16 had achieved PruritusNRS ≥4-point improvement (mMPP)*†‡4
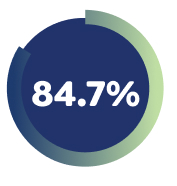
84.7% of Week 16 responders had achieved a Pruritus NRS ≥4-point improvement with EBGLYSS® monotherapy at Week 52 with Q4W.4
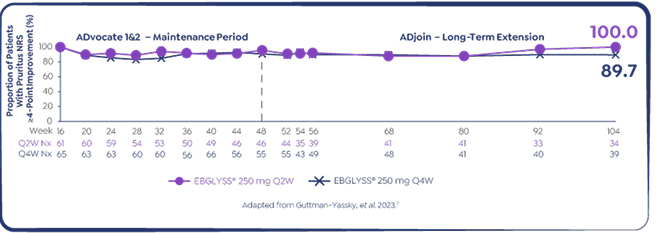
Pooled ADvocate 1&2 and ADjoin study: EBGLYSS® monotherapy at Week 104
Secondary endpoint: maintenance of Pruritus NRS ≥4-point improvement among EBGLYSS® responders7
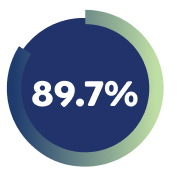
89.7% of Week 16 responders* who had achieved a Pruritus at least ≥ 4-point improvement maintained that response with EBGLYSS® monotherapy Q4W at Week 104 7
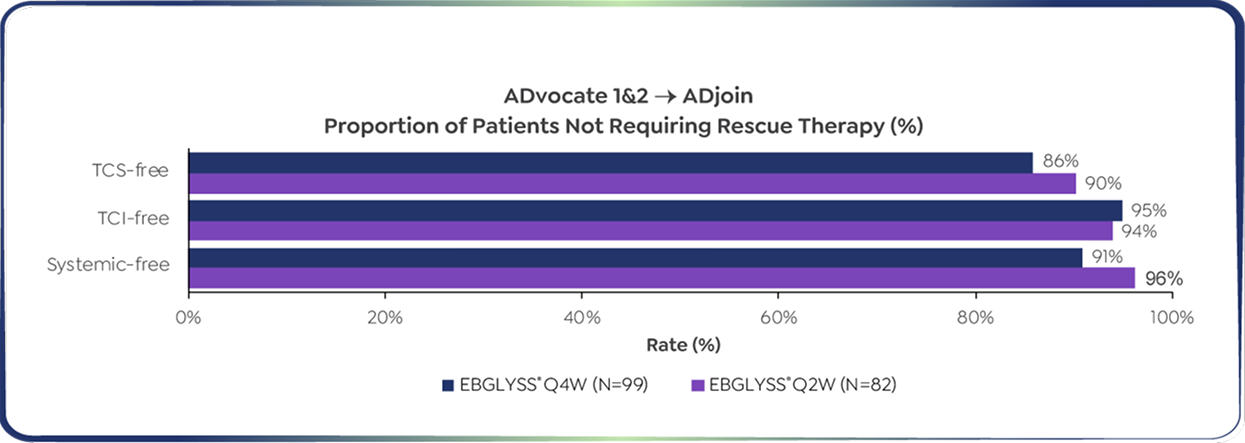
86% of patients did not need TCS throughout 3 years of continuous treatment5
EBGLYSS® has demonstrated a consistent safety profile6
At 16-week the most common adverse reactions are conjunctivitis (6.5%), injection site reactions (0.6%), conjunctivitis allergic (1.8%) and dry eye (1.4%).6
Discover the Clinical Trials: ADvocate 1 and ADvocate 2
▼ This medicinal product is subject to additional monitoring.
FOOTNOTES (EASI-75 AT WEEK 16)
*Patients who achieved EASI-75 at Week 16: ADvocate 1 (N=424): EBGLYSS® (n=283) 58.8%, placebo (n=141) 16.2%; p<0.001; ADvocate 2 (N=427): EBGLYSS® (n=281) 52.1%, placebo (n=146) 18.1%; p<0.001.1,2
†The efficacy analyses in ADvocate 1 were based on the ITT population (which included all the patientswho had undergone randomisation). ADvocate 2 analyses were performed on a modified ITT population, excluding 18 patients (from a single study site) whose eligibility could not be confirmed.2
‡EASI assesses four signs of disease (erythema, papulation or edema, excoriation, and lichenification) across four body regions, with a total score ranging 0 to 72 (with higher values indicating a greater severity and extent of disease). EASI-75 indicates 75% improvement in EASI score from baseline.2,3
FOOTNOTES (EASI-75 AT WEEK 52)
*EBGLYSS® Week 16 EASI-75 responders who maintained EASI-75 at Week 52 with EBGLYSS® Q2W (n=112) 78.4%; EBGLYSS® Q4W (n=115) 81.7%; placebo (EBGLYSS® withdrawal, n=57), 66.4%.4
†Owing to the small number of patients in each dose group from Week 16 to Week 52, analyses for ADvocate 1 and ADvocate 2 used pooled populations that combined the data from both studies. The integrated efficacy analyses were performed on the pooled mMPP.4
‡EASI assesses four signs of disease (erythema, papulation or edema, excoriation, and lichenification) across four body regions, with a total score ranging 0 to 72 (with higher values indicating a greater severity and extent of disease). EASI-75 indicates 75% improvement in EASI score from baseline.2,3
FOOTNOTES (PRURITUS AT WEEK 16)
*Patients who had Pruritus NRS ≥4 at baseline and achieved an improvement of ≥4 points on the 11-point Pruritus NRS at Week 16: ADvocate 1 (N=424): EBGLYSS® (n=283) 45.9%, placebo (n=141) 13.0%; p<0.001. ADvocate 2 (N=427): EBGLYSS® (n=281) 39.8%, placebo (n=146) 11.5%; p<0.001.1,2
†The efficacy analyses in ADvocate 1 were based on the ITT population (which included all the patientswho had undergone randomisation). ADvocate 2 analyses were performed on a modified ITT population, excluding 18 patients (from a single study site) whose eligibility could not be confirmed.2
‡Pruritus NRS: A participant-reported, single-item, daily, 11-point scale used by participants to rate their worst itch severity over the past 24 hours with 0 indicating “No itch” and 10 indicating “Worst itch imaginable”. The minimal clinically important difference is 3 points, while a 4-point change is a more conservative assessment of clinical impact. Pruritus NRS improvement was a secondary endpoint of ADvocate-1 and ADvocate 2.2,3
§A significantly higher proportion of patients in the EBGLYSS® group than in the placebo group had a ≥4-point reduction in Pruritus NRS score at Week 2 in ADvocate 1 (p=0.02) but not in ADvocate 2.2
FOOTNOTES (PRURITUS AT WEEK 52)
*EBGLYSS® Week 16 responders who achieved Pruritus NRS ≥4-point improvement at Week 16 and maintained their improvement at Week 52 with: EBGLYSS® Q2W (n=61) 84.6%; EBGLYSS® Q4W (n=65) 84.7%; placebo (EBGLYSS® withdrawal, n=28), 66.3%.4
†Owing to the small number of patients in each dose group from Week 16 to Week 52, analyses for ADvocate 1 and ADvocate 2 used pooled populations that combined the data from both studies.The integrated efficacy analyses were performed on the pooled mMPP.4
‡Pruritus NRS: A participant-reported, single-item, daily, 11-point scale used by participants to rate their worst itch severity over the past 24 hours with 0 indicating “No itch” and 10 indicating “Worst itch imaginable”. The minimal clinically important difference is 3 points, while a 4-point change is a more conservative assessment of clinical impact.3
FOOTNOTES (EASI-75 AT 3 YEARS)
aEBGLYSS® responders randomized to EBGLYSS® 250 mg Q2W or EBGLYSS® 250 mg Q4W at Week 16 (ADvocate 1&2), and enrolled into ADjoin with the same dosage regimen at Week 52.5
bData from Week 16 responders achieving EASI 75 at Week 16 of parent study.5
cPatients who discontinued treatment due to lack of efficacy had values set to their parent study baseline value subsequent to this time. Observations after discontinuing treatment due to other reasons are set as missing and handled with missing imputation.5
FOOTNOTES (PRURITUS AT 2 YEARS)
*Responders in ADvocate 1&2 were defined as those patients who achieved either EASI-75 or IGA 0/1 following 16 weeks of EBGLYSS® 250 mg Q2W treatment without use of rescue therapy. From a long-term extension study (ADjoin) in responders enrolled from the parent studies ADvocate 1&2 (EBGLYSS® monotherapy) and ADhere (EBGLYSS® + TCS). Only ADvocate 1&2 results are shown. EBGLYSS® Q2W was also studied to Week 104 but is not a recommended dose once clinical response has been achieved.7
REFERENCES
1. EBGLYSS® (lebrikizumab). EU Summary of Product Characteristics.
2. Silverberg JI, et al. N Engl J Med. 2023 Mar 23;388(12):1080–1091.
3. Silverberg JI, et al. N Engl J Med. 2023 Mar 23;388(12):1080–1091. Supplementary appendix.
4. Blauvelt A, et al. Br J Dermatol. 2023 May 24;188(6):740–748.
5. Thaçi D, et al. Efficacy and Safety of Lebrikizumab Is Maintained Up to 3 Years in Patients With Moderate-to-Severe Atopic Dermatitis: ADvocate 1 and ADvocate 2 to ADjoin Long-Term Extension Trial. Presented at the European Academy of Dermatology and Venereology (EADV); Amsterdam, Netherlands; September 25-28, 2024 . D1T01.2.
6. Stein Gold, L., et al. Safety of Lebrikizumab in Adults and Adolescents with Moderate-to-Severe Atopic Dermatitis: An Integrated Analysis of Eight Clinical Trials. Am J Clin Dermatol 24, 595–607 (2023). https://doi.org/10.1007/s40257-023-00792-6
7. Guttman-Yassky E, et al. SKIN The Journal of Cutaneous Medicine, 7(6), s271.